

Michael Hickey/Getty Images Entertainment
Investment Thesis On Amylyx
Amylyx (NASDAQ:AMLX) is a biotech developing a potential therapy for Amyotrophic Lateral Sclerosis (“ALS”) – a devastating disease that leads to “deteriorating muscle function, the inability to move and speak, respiratory paralysis, and death” in a definition supplied by the company itself in a recent investor presentation.
ALS affects ~200k people worldwide, and ~29k people in the US, ~30k people and Europe, and ~3k people in Canada – where Amylyx’s drug AMX0035 is now approved, under the name Albrioza.
I covered most of the background to Amylyx – founded by two Brown University students in 2013, before joining the Nasdaq in January this year via an IPO raising $190m at $19 per share – in a note on the company back in mid June.
I was bearish on Amylyx’ chances of building share price momentum via a US approval of Albrioza, based on the verdict of an Advisory Committee convened by the FDA in March, which voted 4-6 against in answer to the question of whether data from Amylyx’ Phase 2 CENTAUR study proves that the drug is effective in the treatment of patients with ALS.
Nevertheless, Amylyx stock did steadily gain in value in June, July and August – from $18, to a high of $26.5. This likely reflects investor belief that, despite the AdComm verdict, an accelerated approval of Albrioza based on the Phase 2 CENTAUR data remained the likeliest outcome, given the paucity of treatment options for ALS patients.
That optimism has foundered in September, however, with the share price falling back down to $17 at the time of writing – a loss of 36% – after the FDA released briefing documents ahead of a second Advisory Committee, or “AdComm” review of the CENTAUR trial data, due to take place today.
It is an unusual step for the agency to conduct two AdComms in relation to the approval of a single drug, again reflecting its desperation to find and approve an ALS drug that can make a meaningful difference to patient’s conditions.
It shows the agency is taking Albrioza very seriously, but ultimately, its latest briefing notes seem to suggest that it has not found the evidence it was looking for to support an accelerated approval of Albrioza based on Phase 2 trial data.
As such, I’m anticipating another negative AdComm verdict later today and an accompanying loss in the value of Amylyx shares. That does not necessarily mean that Amylyx won’t eventually succeed in ALS with Albrioza, but my expectation would be the FDA will opt to wait for data from Amylyx’ Phase 3 PHOENIX study, which will attempt to enroll up to 600 patients, evaluated over a period of 48 weeks. The study is apparently already 50% enrolled, and ought to be complete by late 2023 / early 2024.
A Phase 3 trial may test Amylyx’ financial resources – the company reported a cash position of $207m as of Q222, and burned through $102m of cash in the first six months of 2022 – and result in at-the-market fundraisings and investor dilution. That will not work out well for shareholders in the short to medium term, but it may be the only way the company will be able to convince the FDA of the efficacy of Albrioza, provided of course that the data is positive.
In this post I will briefly review the FDA’s latest objections to the CENTAUR data, circulated in its briefing notes, and try to evaluate what Amylyx’ next move may be.
Another thumbs down from its AdComm does not necessarily mean that the FDA will not approve Albrioza when its Prescription Drug User Fee Act (“PDUFA”) – originally scheduled for June 29, but postponed to Sept. 29 – arrives, given the AdComm’s verdict is not binding and the FDA can act independently of it.
Given it’s the FDA leading the criticism of the CENTAUR data, and the additional data Amylyx has supplied since, however, even if the AdComm were to surprisingly vote in favor of Albrioza, its eventual approval would still appear doubtful.
Albrioza / AMX-0035 Overview – Drug and CENTAUR Trial
Albrioza is a dual UPR-Bax apoptosis inhibitor composed of sodium phenylbutyrate (“PB”), and TURSO – also known as tauroursodeoxycholic acid, or TUDCA. Amylyx says that it has shown in multiple models that the drug:
can keep neurons alive under a variety of different conditions and stresses, including in in vitro models of neurodegeneration, endoplasmic reticulum stress, mitochondrial dysfunction, oxidative stress and disease-specific models of a variety of other conditions, as well as in vivo models of Alzheimer’s Disease, or AD, and multiple sclerosis.
AMX0035 mechanism of action explained. (investor presentation)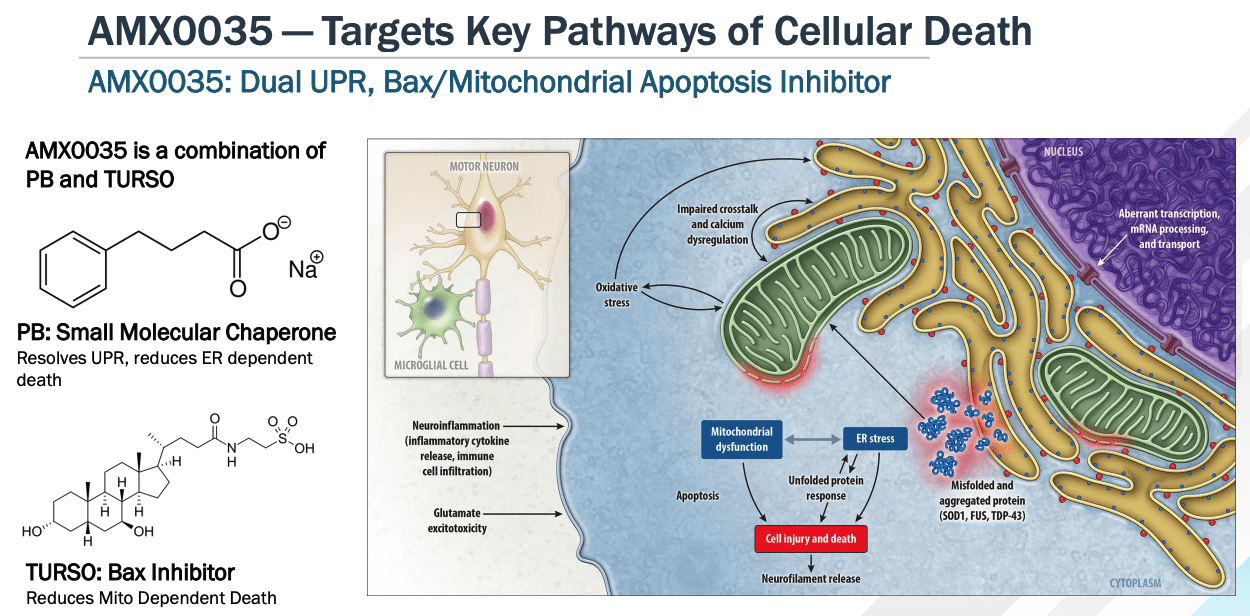
As we can see above, the dual components of AMX0035 target unfolded protein responses (“UPR”) and the mitochondrial Bax pathway – key mediators of cell death, as per the Nobel Prize winning (in 2002) work of scientists Sydney Brenner, Distinguished Professor at The Salk Institute in La Jolla, California, Robert Horvitz, Professor of Biology at MIT, Cambridge, MA, and Sir John Sulston, former Director of The Wellcome Trust Sanger Institute, in Cambridge, UK.
The Phase 2 CENTAUR study evaluated 137 ALS patients with slow vital capacity (volume of air expired through an unforced manoeuvre after maximum inhalation) >60%, with Riluzole / Edaravone (other ALS therapies – discussed below) use permitted, and a 24-week randomisation phase between placebo and drug.
The results demonstrated a statistically significant, 2.32 point difference (p=0.03) in ALS Functional Rating Scale (“ALSFRS-R”) scores – considered a gold standard assessment of patients conditions, being a 5-minute questionnaire using an ordinal rating scale of 0-4, covering 12 functional activities relevant to ALS across four functional domains – between the patient and placebo arms.
Treatment with AMX-0035 also resulted in longer lifespans for patients using the drug – a 6.5 month difference – according to the study. Later analysis suggested that randomization to AMX0035 resulted in 10.6 months longer median survival duration using the rank-preserving structural failure time model (“RPSFTM”).
The FDA Responds To Data – Old and New
I summarized the FDA’s initial response to Amylyx’s CENTAUR data in my previous note as follows:
Th FDA called the statistical evidence – a treatment difference of 2.32 on the ALSFRS-R scale (p = 0.034) “not highly persuasive,” questioned the “linearity of ALSFRS-R over time assumption” used to prove statistical significance, and highlighted missing data – 17% on placebo/18% on the drug were alive but missing ALSFRS-R Total Score values at Week 24.
The agency also noted that patients also were using the approved therapies edaravone or riluzole during the trial, which could lead to biased results, and also pointed to a “randomization implementation problem” which saw the first 18 patients in the trial assigned to the drug arm.
These and other criticisms – such as the study’s failure to meet any of its secondary endpoints, including Accurate Test of Limb Isometric Strength (“ATLIS”), Levels of plasma Neurofilament heavy chain (“pNF-H”), or Slow Vital Capacity (“SVC”), a key measurement of pulmonary capacity, likely counted against Amylyx and persuaded the AdComm not to back the drug.
So far, not so good, however Amylyx rapidly responded to the FDA’s objections with new data and analysis. Unfortunately, in its briefing notes, the agency appears to be unconvinced by any of the new data.
The first set of data was an individual responder analysis which compared:
the progression rate of individual subjects during the study to their own progression rates prior to entering the study as independent confirmatory evidence of individual effect of the treatment.
This was an attempt to show the FDA that a greater proportion of patients showed an individual response in the AMX0035 arm than in the placebo arm, but the FDA was unmoved, describing this data as “highly correlated with the primary analysis,” and concluding “it does not appear that this data can be considered independent confirmatory evidence as it uses the same data as the primary analysis.”
Next, Amylyx argued that because the majority of placebo patients (71%) crossed over into the Open Label Extension part of the trial, “the Intent To Treat overall survival does not account for treatment crossover and may underestimate the survival benefit of the drug.”
As a result, Amylyx provided two post hoc sensitivity analyses using two different approaches – Natural History data, and a Rank Preserving Structural Failure Time Model (“RPSFTM”). The FDA was apparently persuaded by neither, however, criticizing the Natural History data algorithm for being a “non-randomized comparison to an external control that is subject to potential confounding,” and describing the RPSFTM data as “not independent data” and “simply a new method for analyzing the same survival data presented in the original NDA submission.”
Amylyx also attempted to use data from a clinical trial of AMX0035 in Alzheimer’s where certain CSF biomarkers supported the “mechanistic activity of AMXOO35 in the central nervous system.” The FDA responded that “there is no clear or consistent relationship between the biomarkers that had nominally significant findings and ones that did not.”
There was further discussion of the data which I would encourage prospective investors or shareholders to study in full, but the gist of the FDA’s briefing documents seems to hinge on its conclusion that, in order for a drug to qualify for an accelerated approval – which means that it is approved while pivotal studies are still ongoing, due to the critical level of unmet need in the patient population:
an effect on an endpoint supporting accelerated approval must be an effect on an endpoint that in its character is reasonably likely to predict clinical benefit
In the case of AMX0035, the FDA writes in its briefing notes:
we do not have data for an effect of AMX0035 on an endpoint that is reasonably likely to predict clinical benefit for ALS. The ALSFRS-R and survival are direct measures of clinical benefit and are acceptable endpoints to support traditional approval; therefore, if the Applicant provided data for these endpoints that met the substantial evidence requirements, the Agency would be able to grant a traditional approval.
Certainly, the above statement seems to suggest that the FDA has already concluded that accelerated approval is not appropriate for AMX0035. It will be up to the AdComm to disagree with these conclusions, after hearing from both Amylyx – backed by numerous patient advocacy groups – and the FDA, today.
ALS Treatment Market Overview
Current treatment options for ALS patients are as follows:
- Rilutek – a generic name for Riluzole, approved in 1995 and designed to inhibit glutamate release, prolonging life by ~3 months
- Tiglutik, or thickened Riluzole, approved in 2018 and designed to be easier for patients with declining health to swallow
- Exservan, an oral film formulation of riluzole approved in 2019
- Radicava, or edaravone, which targets unstable molecules thought to contribute to neurodegeneration and was approved in 2017, with an oral version approved in May this year.
- Nuedexta, approved to treat Pseudobulbar affect (“PBA”), a symptom of ALS characterised by frequent, involuntary, and sudden episodes of crying.
Radicava – originally a treatment for Stroke approved in Japan – was approved in ALS in the US in 2017, based on a Phase 3 trial conducted in a subset of patients with greater baseline functionality. Data showed that patients with a forced vital capacity (“FVC”) – the maximum amount of air you can forcibly exhale from your lungs after fully inhaling – of >80% achieved a statistically significant improvement in the ALS Functional Rating Scale (“ALSFRS-R”).
Other studies have concluded that Radicava – marketed and sold by Mitsubishi Tanabe, with a list price of ~$170k – demonstrated no clinical benefit, which tells us two things – that ALS is an area of high unmet need, and that the bar for an ALS drug approval is not high – any drug that can demonstrate even the faintest improvement in patient’s conditions is likely to be approved quickly.
An oral version of Radicava was approved in May this year, which will help Mitsubishi Tanabe circumvent a 2024 patent expiry. Although it’s not expected to improve upon the efficacy of IV Radicava, it ought to help the drug generate sales of >$300m per annum, analysts believe.
Conclusion – Accelerated Approval On Sept. 29 Looks Unlikely and Journey To Traditional Approval Looks Tricky
Central Nervous System disorders, notably the likes of Alzheimer’s, Parkinson’s, and ALS, are notoriously hard to treat – and the FDA made a major misstep in June last year when approving Biogen’s (BIIB) Aduhelm to treat Alzheimer’s, despite inconclusive trial data and dangerous side effects which included brain swelling.
The backlash against that approval was so strong – and the CMS’ refusal to provide reimbursement for patients’ using the drug, except within a clinical trial setting so devastating – that Biogen has all but abandoned Aduhelm as a product, and has moved on to a new Alzheimer’s therapy.
The FDA was in fact supportive of Biogen and the evidence presented for Aduhelm during its AdComm, whilst members of the AdComm voted 10-0 against approval. The situation with Amylyx and AMX0035 is almost the reverse, with the FDA sceptical that the biotech’s data supports and accelerated approval, whilst the AdComm is split.
Should that give investors fresh hope? I would be doubtful on that front, since the FDA is clearly desperate to avoid another controversial approval for a CNS drug, and does not have to follow the decision of its AdComm anyway, even if they’re more persuaded by AMX0035 than the agency itself.
The evidence heard today by the AdComm will be emotive – ALS is a devastating disease and patients and their families are desperate to find a cure – but it has to be said that AMX0035 is certainly no miracle cure for the disease, and even if it were approved this September, its capacity to alleviate patient’s suffering seems extremely limited based on the evidence to date.
What intrigues me most about the new data in relation to AMX0035 is the longer median survival data – up to 18.8 months according to post hoc analyses conducted by Amylyx. In response to this data, the FDA comments that there was “no pre-specified protocol and/or analysis plan for this comparison, and post hoc analyses are challenging to interpret.”
The agency clearly believes that there are too many assumptions made to generate this data, such as use of patient data not related to the CENTAUR trial, and also points out that “death alone was not a pre-specified primary or secondary endpoint in the double-blind period or the OLE.”
I wonder if the AdComm will view this apparently promising data more favorably – such an extension of survival duration in patients seems to provide a compelling reason to introduce the drug sooner rather than later – but with that said, prescribing physicians may not be convinced by the evidence presented and refuse to recommend the drug to patients. CMS may also refuse to reimburse what would likely be a >$150k list price.
In summary, what’s needed is more persuasive, less contrived data – almost impossible to achieve when testing CNS drugs – but on this occasion I suspect Amylyx will be tasked with that goal, and then the challenge will be not just one of drug development, but also of financial management.
It may take Amylyx an extra 18 months and $200m of funding to achieve its approval goal, but the impact the drug may be able to have if the additional data supports the initial optimism could be Amylyx’ saving grace.
Be the first to comment