

Tharakorn/iStock via Getty Images
We are short Akero Therapeutics (NASDAQ:AKRO) because we believe investors are not getting the full picture from the company’s approach to presenting the data from its clinical programs. Add to this that the drug has other major liabilities in terms of both its poor tolerability and the fact that it requires regular injections. Moreover it will be competing in against a very large number of competitors and will need to outperform them all. This is compounded by the fact that despite the seemingly large cash balance of $380 million (pro forma), we believe that this is unlikely to be sufficient for the company to finance the large Phase III clinical program that will be needed to potentially bring this product to market.
Company overview
The company is developing the drug efruxifermin (EFX) for the treatment of nonalcoholic steatohepatitis (NASH). EFX is a drug that mimics the protein fibroblast growth factor 21 (FGF21), and the hope is that this activity can improve the accumulation of fat and the formation of fibrotic tissue that occurs in the liver when a patient has NASH. The thesis is reasonable as this is a drug that targets pathways involved in fibrosis. Indeed, this isn’t the first time that a drug targeting FGF21 has been developed, and indeed a selection of Big Pharma companies have developed and discontinued their own drugs of this class: pegbelfermin (BMY), NN9500 (NVO), aldafermin (MRK), and RG7992 (OTCPK:RHHVF) are all examples. LLY and PFE also developed FGF21 inhibitors which were tested in diabetes (but not NASH), neither of which progressed in clinical trials. EFX is somewhat different than these older products in that it functions as a mimic of FGF21 as opposed to directly targeting the molecule’s receptor.
The company licensed the drug from Amgen (AMGN) for $5 million and 10% of its outstanding shares at the time (5.8 million). They’ve subsequently made a $2.5 million milestone payment to AMGN and owe them an additional $7.5 million in the event that the product has positive Phase III results (as well as $105m in commercial and regulatory milestones). This is exceptionally cheap, and we have little information regarding why AMGN chose to divest the asset.
The company currently has no other products in the clinic and no other development programs it has provided any details on, so the entirety of the thesis rests on the activity of EFX.
Missing in action
In September 2022, The company presented some data with a very rosy picture from its recently completed Phase IIb study. However, these data like those that were gathered in prior clinical trials omit multiple patients and do not follow the so called “intent-to-treat analysis” (ITT) conventions, which is a standard criteria for drug approval (pg. 28, among much of the text). Deviations from ITT are only accepted when that type of analysis cannot be performed, but there are no such limitations here. Decades of approvals are consistent with this policy. A so called modified ITT (mITT) can be used at times and at the discretion of the FDA, which allows for the exclusion of patients that had no data gathered on them, although this term isn’t used completely consistently. To be clear, that is not what AKRO did here. They excluded patients that withdrew because of a range of reasons including side effects and excluded patients that completed the study.
The FDA requires ITT data because patients discontinuing treatment can very strongly bias the data. Patients discontinue for a range of reasons, which can include intolerability of the drug, a lack of perceived benefit, the need for a different intervention, etc., but these patients almost by definition are unlikely to be those with very strong clinical responses to the drug. However, when these patients are excluded from the analysis, this causes the opposite effect of increasing the reported effect size. If hypothetically a drug worked in 10% of patients, but the other 90% couldn’t tolerate the treatment and withdrew from the study, this does not mean that the drug has a 100% response rate.
The data that the company presented effectively doubled the company’s market cap. These data showed marked improvement in fibrosis and liver fat, including the holy grail endpoint of improvement in both at the same time. This would be fantastic, except we find that different numbers of patients are included in the different analyses. For instance when discussing baseline characteristics there are 43 patients in the high dose (50mg) arm, but when discussing the primary endpoint, 9 of these patients have disappeared. That’s 21% of all the data available for this arm. Even among the endpoints being presented, there is no consistent count of patients, with them being 34, 35, or 36 depending on the slide when looking at efficacy, but looking at the full population of 43 when looking at the rate of side effects.
Akero Akero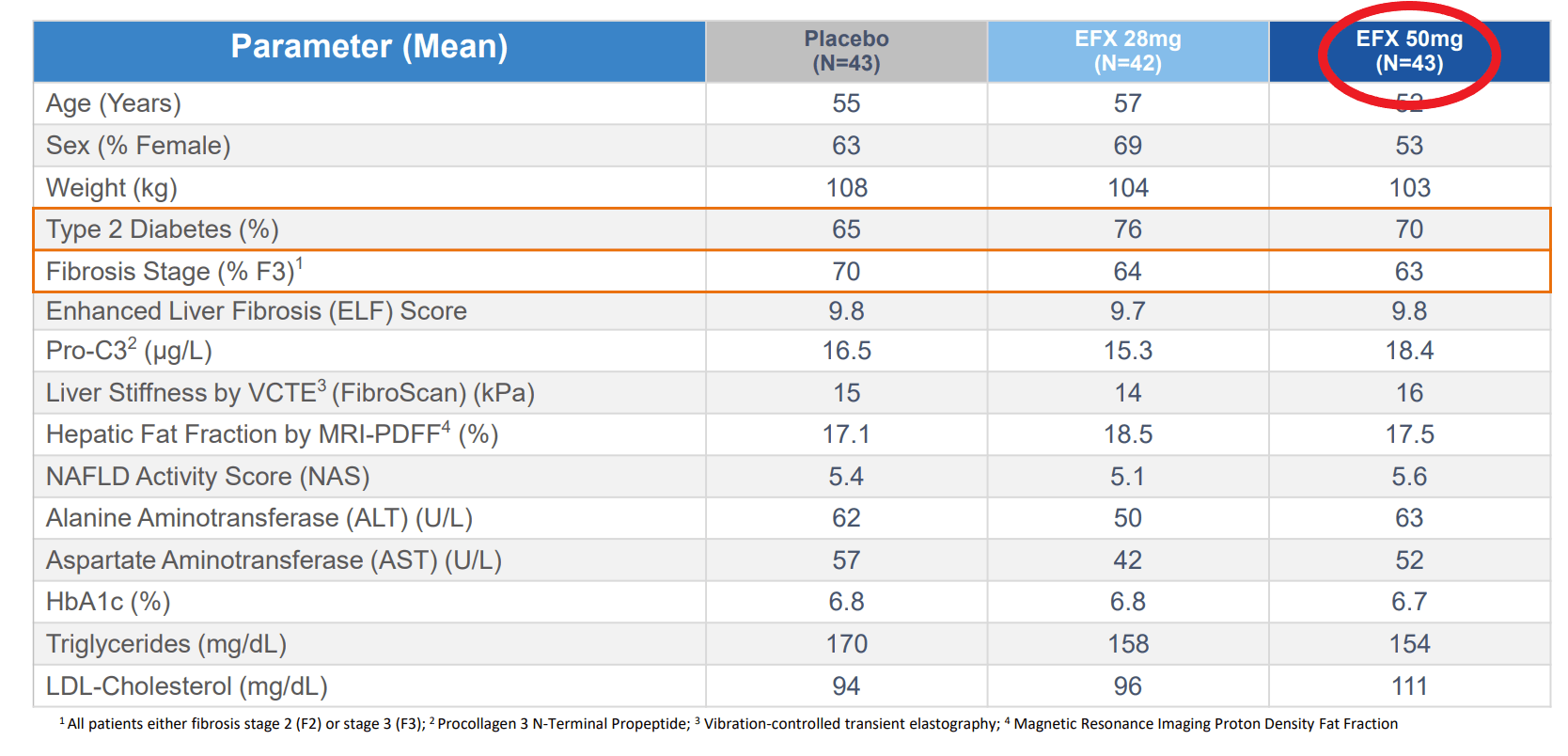
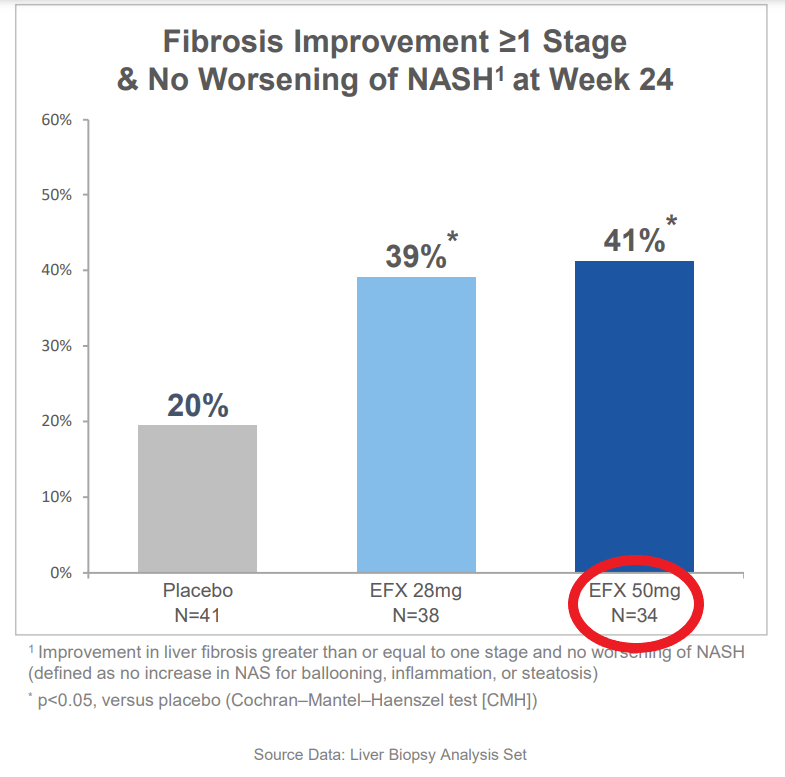
The reason for this discrepancy was that only patients that opted for liver biopsies were included in the results. Most of the patients that completed the study agreed to have a biopsy taken. Rather the majority of these missing patients are ones that did not complete the treatment course.
To be clear, there is no confusion regarding this data, as the company has been directly asked about the differences in these patient numbers between different analyses during the conference call in which they were presented (conversation starting at 43:30). First, the company provided an explanation for why they chose not to do an ITT analysis, which was essentially that other companies exploring NASH have similarly not presented ITT results. This is certainly true, and we would have the same criticisms, but it is by no means universal.
The CEO Andrew Cheng stated:
“I think we lose the 50mg arm on an ITT basis, and you can see why that makes sense given the slightly higher number of dropouts in that.”
He is referring to the primary endpoint of improvement in fibrosis without worsening in NASH. So the trial would have at least missed the primary endpoint at the highest dose. He also stated that measure of fibrosis would come down across the board for all the reported endpoints, regardless of significance.
This also isn’t the first time that AKRO has presented data only on a section of patients from their studies. The company’s prior Phase IIa data similarly didn’t include patients for a wide range of reasons. For instance only 2 patients in that study’s placebo arm were included in the efficacy analysis. Almost 50% of all the patients in the study were screened from the analysis.
Akero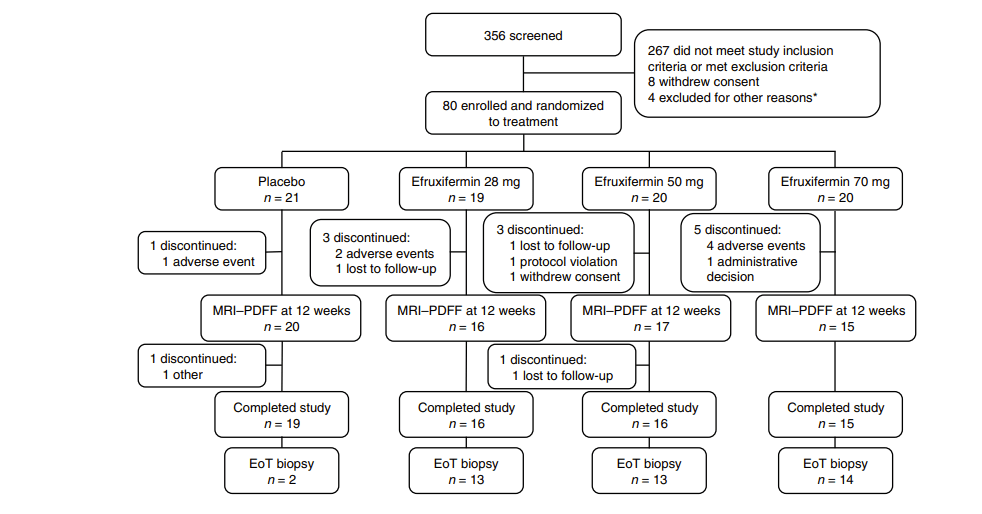
Not the only limitation in the data
In addition to the above questions regarding the quality of data presented, some questions were also raised regarding the tolerability of this compound. 33-35% of people on the drug arms had diarrhea and another 25-33% had nausea. Before even considering ITT, these are high rates for a drug you would be expected to take chronically for years or the rest of your life.
Another previously known liability of this product is that it must be injected, which also limits the applicability of a product intended for long term use. The vast majority of other products in development for NASH are oral, so it may be hard to compete with a product that is harder to administer.
Because of these limitations, may be more attractive to desperate patients with few options, namely patients with advanced (F4) fibrosis, as opposed to moderate (F2-F3) like in the studies described above. The moment of truth for this program may be with the readout of the company’s ongoing Phase IIb trial in F4 patients, which is expected to read out in H223. The F4 indication is substantially harder than earlier stage disease, and there has been little if any success in treating this indication.
A long way to the finish line
The future of this company is tied up in these programs. It doesn’t have any other programs in development that have been disclosed. The company raised cash ($200 million) immediately after the Phase IIb data, leaving them with a pro forma balance of $380m, or about 3.5 years at the current run rate (-$110m EBITDA), but this is before considering the sheer scale of what a Phase III program looks like in NASH.
NASH studies are very long and costly. For instance, Intercept Pharmaceuticals (ICPT) recently reported results from its Phase III NASH study, which had been going on since 2017. Madrigal (MDGL), is expected to present data at years end from their 2000 patient study, which is in addition to their 3 other ongoing Phase III clinical trials, with enrolment from 700 to 1400. Madrigal completed its Phase II studies in 2018, and initiated its pivotal studies in 2019.
MDGL will be presenting its final pivotal data in Q4 2022, and its timeline gives an idea of what AKRO’s could look like. It’s been 5 years since they initiated the above study in 2017, and there will be an additional year of FDA review before approval. It may be possible for AKRO to run a faster study than this but we believe a three year study and the statutory 1 year of review is probably optimistic, putting approval in 2026 at the earliest.
AKRO spent $89 million on R&D in the past 12 months. This was to support the above 128 person clinical study. You can envision how large this R&D cost can get when running a study that’s over 15 times the size, like MDGL’s ongoing pivotal study. MDGL’s R&D expense has been $214 million over the past 12 months, and we expect AKRO’s spending to look similar. We therefore think it is unlikely that the company will be able to reach approval even with its current large cash balance. If we extrapolate from MDGL’s spending, AKRO has about a year and a half before they need more cash, and they haven’t even started their pivotal studies yet.
Also during the time that this product is in Phase III studies, there will be numerous reports on other drugs for NASH, which the current data will be compared against. There are an hundreds of drugs in development and ongoing clinical trials for NASH.
Valuation
We don’t believe that AKRO will ever be a cash generating business, so we believe a fair valuation is well below cash. We can expect sequential dilution in the interim. At the current share price, one year of spending at MDLG levels (approximately $250 million in negative EBITDA) corresponds to 14% dilution, and we can expect these studies to take at least three years, potentially longer, and then the company will need to wait through an FDA review for an additional year.
The company is currently highly valued at approximately $1.2bn in market cap, of which about a third is cash. This is a very high valuation for this early stage and implies that the market is considering a high probability of success as well as high sales. It is comparable to MDLG in size, despite the latter having imminent Phase III results and a potential near-term approval. It is one of the most highly valued of any NASH company, despite its early stage. Any slippage in this market valuation will substantially increase the level of dilution needed, and this effect will compound through multiple rounds.
A catalyst for the stock will be when the company presents more data from this program at an upcoming medical conference. The company may present a more complete picture at this time (although that cannot be counted on). Additionally, the final data from MDGL’s pivotal study is expected before the end of 2022, and if it is good, this may negatively impact AKRO.
Risks to the thesis
The main risk to the short thesis here is that the company’s drug is stellar and becomes the market leader in NASH. We are not particularly worried about this risk for two reasons:
- The above described limitations make us lack confidence in the approvability and commercial potential of the product
- The dilution that the company is facing in order to complete its clinical trials makes the stock actionable in the nearer term.
In the years that it will take to complete the current program, there will be numerous announcements from other companies developing products for NASH, and we believe that this will lead investors to reconsider the attractiveness of EFX and the data being presented now.
Another risk is that the company is acquired, but we believe that this is currently limited by the company’s high valuation. In addition, the business development teams at the Big Pharma companies that would consider this transaction are going to get to see all the data under NDA.
Moreover, the trade caries the typical costs and risks of a short. The borrowing rate is inexpensive in the low single digits, but this might increase if more people short the stock. An alternative trade involving puts has the same general principle, although options are more susceptible to changes in short term volatility, which can be high in catalyst generating stocks.
Conclusion
We believe that NASH is a very hard space to do drug development in and that AKRO is doing its best develop this drug. However, the company has chosen not to provide the complete data from its clinical studies multiple times. The company admitted that this would have caused them to miss a key endpoint. Without the type of data the FDA needs, it is hard for investors to draw conclusions regarding the eventual approvability of this product.
These data they did present, in this case, has had a dramatic impact on the stock. We cannot deny that. This is one of the most highly valued companies in NASH. However, we believe that these gains will be short lived as the company deals with the reality of running Phase III studies on such a drug, and as the market is exposed to the countless other NASH programs from other companies, some of which might present ITT data to their investors.
Be the first to comment